









单胺类神经递质是广泛分布在人体内的一类化学信号分子,包括多巴胺(dopamine, DA)、肾上腺素(adrenaline)和五羟色胺(serotonin, 5-HT)等,这些信号分子共同调控人体内包括情绪以及记忆在内的多种生理功能并维持机体内环境稳态。多巴胺作为人体内一种重要的单胺类神经递质,通过多巴胺能神经系统,对中枢神经系统(CNS)以及外周神经系统(PNS)的功能进行调控。多巴胺能信号主要由人体内一类被称为多巴胺受体(dopamine receptors, DRs)的G蛋白偶联受体(G protein-coupled receptor, GPCR)介导,包括D1R到D5R共五个受体成员。按照偶联下游G蛋白种类的不同,这些受体可以进一步分为D1类受体和D2类受体两组。其中,D1类受体包含D1R和D5R,主要与激活型G蛋白Gs偶联,刺激下游第二信使环状单磷酸腺苷(cAMP)的生成,而包括D2R,D3R和D4R在内的D2类受体则主要与抑制性G蛋白Gi/o偶联,抑制cAMP形成。在五种DRs中,D1R和D2R是CNS中表达最为丰富的受体,主要分布在基底神经节和前额叶皮层中 1,2。D1R和D2R介导的多巴胺能信号对于奖赏、认知、运动协调和神经内分泌功能等在内的高级脑部功能至关重要,其发生异常与许多神经精神疾病密切相关,包括阿尔兹海默症(Alzheimer's disease, AD)、帕金森氏病(Parkinson’s disease, PD)、精神分裂症、认知障碍、注意力缺陷多动症(Attention deficit hyperactivity disorder, ADHD)以及药物成瘾和滥用等。作为多巴胺受体家族的代表成员,D1R和D2R是治疗PD以及精神分裂症的热门药物靶点。
(图源:https://www.cerveauetpsycho.fr/sr/cerveaux-confines/8-la-dopamine-recompense-du-confinement-19233.php)
D1R选择性激动剂长期以来被认为是治疗PD的有效方法,然而,目前上市的D1R激动剂药物大多为D2样受体的选择性激动剂。已经开发的D1R选择性激动剂由于受到代谢快(儿茶酚结构特征)以及无法透过血脑屏障等缺陷影响,尚无通过临床研究用于神经精神类疾病治疗 3;在治疗精神分裂症中,尽管可以通过目前的药物作用D2样受体来有效地治疗正面症状,但它们在减轻负面症状和认知缺陷方面效率低下,而D1R选择性激动剂则可以作为精神类疾病患者提高认知的潜在治疗途径。此外,D1R激动剂也被普遍认为是治疗AHDH以及药物成瘾的有效治疗方法。通过对D1R和D2R受体进行结构药理学研究并揭示其配体选择性的分子机制,对理解配体结合特性、受体激活以及设计更为高效的多巴胺受体靶向抗神经精神疾病类药物具有重要的科学意义和临床应用价值。
目前,尽管已有若干多巴胺受体亚型的结构获得解析,包括D2R,D3R和D4R与拮抗剂结合复合物的晶体结构,以及D2R(突变型)与激动剂复合物的低分辨率冷冻电镜结构,然而,对于D1类受体,尤其是D1R,自D1R基因被发现及克隆近30年来,其受体结构仍处于未知状态,极大地限制了人们对D1R配体识别和受体激活机制的理解,成为制约基于结构的靶向D1R受体药物研发的重要科学瓶颈。
针对以上科学难题,中美两国科学家联合攻关,来自中国科学院上海药物研究所徐华强课题组和浙江大学基础医学院与浙江省良渚实验室张岩课题组,联合美国匹兹堡大学张诚课题组以及北卡罗来纳大学教堂山分校Bryan L. Roth课题组等,应用冷冻电镜技术(Cryogenic electron microscopy, Cryo-EM)首次解析了帕金森病治疗药物apomorphine(多巴胺受体泛激动剂)、D1R/ D5R选择性全激动剂SKF81297以及G蛋白信号偏好性D1R/D5R选择性部分激动剂SKF83959激活下D1R与下游Gs蛋白复合物的高分辨率冷冻电镜结构,分辨率为2.9 Å - 3.0 Å。同时,科研人员解析了帕金森病治疗药物bromocriptine激活下D2R(野生型)与Gi复合物2.8 Å分辨率的冷冻电镜结构(图1)。这些结构数据结合功能实验结论,揭示了D1R和D2R配体结合口袋的拓扑结构特性、受体激活机制、激动剂选择性识别并激活D1R和D2R的分子机制、D1R的G蛋白偏好性激活决定因素以及D1R和D2R在G蛋白选择性差异的结构基础等。
以上研究成果为以D1R和D2R为药物靶点的选择性激动剂药物的设计和开发,以及G蛋白信号偏好性D1R靶向药物设计提供了重要的结构基础和理论依据。研究论文“Structural insights into the human dopamine D1R and D2R receptor signaling complexes”,以长文形式于2021年2月11日在国际顶级期刊Cell杂志上在线发表。这是继2月5日发表在Molecule Cell上D3R的工作之后徐华强课题组和张岩课题组在多巴胺能系统方向进行结构和功能系列研究的又一突出研究进展,进一步加深了人们对该系统的认识。
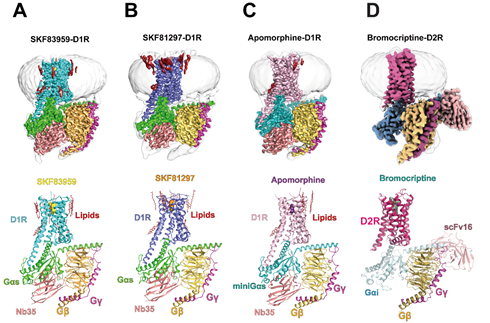
图1. D1R-Gs和D2R-Gi复合物结构
研究发现,D1R在结构上表现为经典的七次跨膜螺旋结构,其中配体正构结合位点位于受体胞外端,由胞外loop以及跨膜螺旋部分组成;Gs蛋白偶联界面位于受体胞内端,由近胞内端结构域组成。SKF81297、SKF83959以及apomorphine都属于D1R的儿茶酚胺类激动剂,在与D1R的结合上,三种激动剂与受体上结合口袋的相互作用模式类似,其中最为典型的是配体上的氨基与D1033.32之间形成离子相互作用,这个作用位点在所有单胺类受体上及其保守。在结合模式上,三种激动剂的儿茶酚结构朝向TM5,但整体构象存在细微差别,而这种细微差别导致了不同配体在激活效力以及信号转导通路偏好性的差异。通过对比D1R-SKF81297、D1R-SKF83959的结构细节,研究团队发现,虽然SKF83959仅仅比SKF81297多了两个甲基,然而,受到SKF83959结构上azepine环上额外的甲基与D1R上疏水氨基酸F3137.35、W3217.43空间位阻效应影响,相比于SKF81297,SKF83959更接近TM5并限制了TM5向跨膜区中心内移,使得SKF83959表现出比SKF81297更弱的激活效力,这与salmeterol部分激活β2AR的原理相似。此外,SKF83959作为D1R的G蛋白偏好性激动剂的机制长期以来始终未得到解释,通过结构比对和β-arrestin募集实验分析,研究团队发现与SKF83959上azepine环内额外的甲基相互作用的D1R TM5上的氨基酸残基F2886.51、F2896.52以及TM7上的V3177.39在SKF83959的G蛋白偏好性活性上起重要作用,为设计更为安全的G蛋白偏好性D1R激动剂提供了重要的结构基础和理论依据。
研究人员对比D1R与非选择性激动剂apomorphine的结构发现,apomorphine的结合比SKF化合物更远离D1R的胞外loop 2(ECL2)。进一步比较D1R和D2R的结构,研究团队发现D1R和D2R的ECL2在拓扑结构上存在明显的差别,D2R的ECL2上的“CIIA”基序相比于D1R的“CDSS”基序更为靠近正构结合位点中心,如果SKF化合物和apomorphine以类似的模式结合到D2R,D2R的ECL2,尤其是氨基酸残基I184,将与SKF化合物而不是apomorphine发生空间位阻效应。比较D2R-bromocritine的结构发现,bromocriptine远离D2R ECL2区域,避免了与之发生位阻效应,这些结果表明ECL2在D1R和D2R的配体选择性上发挥重要调控作用。Bromocriptine在对D1R的结合力上比D2R大概低50倍。通过D1R和D2R的结构比对发现,D1R的配体结合口袋更为狭窄,相比于D2R,D1R的TM6近胞外端往跨膜中心内移5.5 Å并于bromocriptine发生一定程度空间位阻,D1R上的非保守的带正电氨基酸K81也在能量上不利于和bromocriptine的结合,这些因素共同决定了bromocriptine对D2R更高的亲和力(图2)。
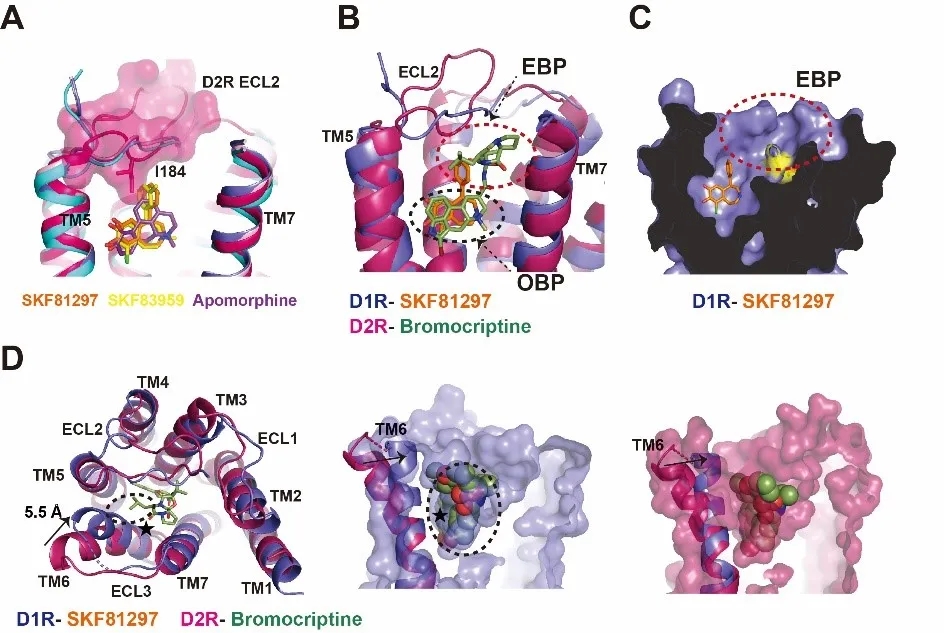
图2. D1R和D2R配体结合特性的比较
虽然D1R与D2R分属于同一GPCR家族,然而在进化树分析上D1R与β2AR更为接近。与之对应的是,在结构上,D1R和β2AR表现出高度的相似性,尤其是在跨膜区TM5-7,保守的P5.50I3.40F6.44基序以及DR3.50Y基序上,这些发现也预示着D1R和β2AR具有相似的激活机制。同为单胺类受体,D1R和β2AR表现出对不同单胺类神经递质的选择性。研究团队发现,将D1R TM7上的V317突变成β2AR对应的氨基酸残基天冬酰胺(N),能显著性提高D1R对β2AR选择性神经递质肾上腺素及其衍生物异丙肾上腺素的结合效力,表明V3177.39在决定D1R对多巴胺而非其他单胺类神经递质的选择性上至关重要。
在下游G蛋白偶联上,D1R主要偶联到Gs,而D2R主要偶联Gi。科研人员同时对两者偶联下游G蛋白选择性的机制进行了探索。结果发现,激活态下D1R和D2R的近胞内端结构的构象差异直接引起各自在偶联下游G蛋白的不同(图3),这些差异体现在以下几个方面:①. D1R TM6的近胞内端相比于D2R外移了8.4 Å,以容纳Gαs上α5螺旋C末端庞大的氨基酸侧链,而D2R近胞内端区域形成的凹腔不足以容纳Gαs 的C末端复杂的氨基酸侧链并与之发生空间位阻,相反,却可以容纳Gαi的C末端较为简单的氨基酸侧链,从而导致D1R和D2R对不同G蛋白的选择性;②. D1R的TM5相比于D2R较长,往胞内区更多延伸了2.5个α螺旋并于Gαs的Ras结构域形成进一步的相互作用;③. D1R的ICL2相比于D2R多了1个α螺旋,使之与Gαs的Ras结构域形成更强的疏水相互作用网络。这些因素表明TM6和α5螺旋等的构象决定着D1R和D2R对Gs/ Gi的选择性,这也与徐华强研究员团队在2018年报道的Rhodopsin-Gi结构通过分子动力学模拟所揭示的Gs/Gi选择性机制相符合4。
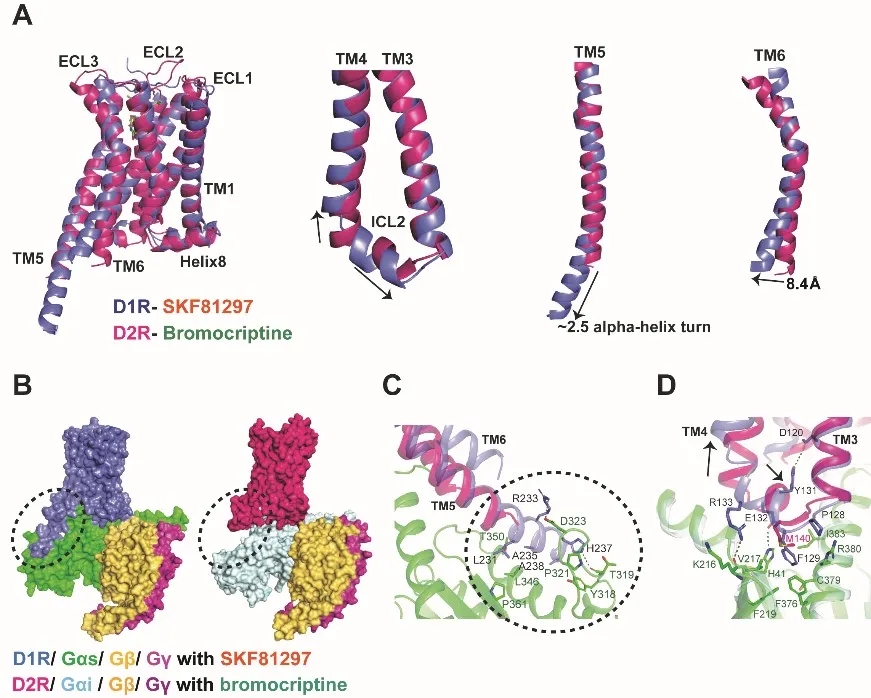
图3. D1R和D2R在G蛋白偶联上的差异比较
综上所述,研究团队通过解析选择性D1R激动剂以及非选择性多巴胺受体激动剂激活下D1R-Gs以及D2R-Gi复合物的结构,结合功能试验数据,阐释了D1R和D2R在配体选择性以及G蛋白选择性识别上的机制等重要的生物学问题,为开发以D1R和D2R为靶标的选择性药物以及更为安全的抗神经精神疾病类药物提供了重要的结构和理论基础。
同时,2月11日同期Cell上,来自四川大学的邵振华团队和北京大学的孙金鹏团队以“背靠背”形式发表了“Ligand recognition and allosteric regulation of DRD1-Gs signaling complexes”的研究论文,该研究报道了D1R与不同激动剂配体以及D1R与变构调节剂的结构,揭示了D1R的激动剂配体以及变构调节剂的结合特性以及潜在的变构调节机制。值得一提的是,针对D1R与内源性配体dopamine以及正向变构调节剂的的结合以及调节机制这一科学问题,徐华强课题组、张岩课题组以及Bryan L. Roth课题组也开展了进一步的研究,相关成果已经以预印本形式在线发表在BioRxiv网站上(https://www.biorxiv.org/content/10.1101/2021.02.07.430101v1)。
本研究冷冻电镜数据在上海药物所冷冻电镜平台以及浙江大学冷冻电镜中心收集。上海药物所2020届博士毕业生庄友文、上海药物所博士生徐沛雨、浙江大学基础医学院博士后毛春友、美国匹兹堡大学博士后Lei Wang、北卡罗来纳大学教堂山分校Brian. Krumm以及美国温安洛研究所X. Edward. Zhou为该论文的共同第一作者。徐华强研究员、张诚教授、张岩教授以及Bryan L. Roth教授为共同通讯作者。上海药物所为本研究第一完成单位。研究工作同时得到了上海药物所蒋华良院士和美国温安洛研究所Karsten Melcher教授的支持和帮助。该工作获得了上海市市级科技重大专项、科技部重点研发计划、中科院先导项目、国家自然基金委、浙江省自然基金委以及美国国立卫生研究院等的项目资金资助。
文章链接:https://doi.org/10.1016/j.cell.2021.01.027
参考文献:
1. Beaulieu, J. M. & Gainetdinov, R. R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacological reviews 63, 182-217, doi:10.1124/pr.110.002642 (2011).
2. Missale, C., Nash, S. R., Robinson, S. W., Jaber, M. & Caron, M. G. Dopamine receptors: from structure to function. Physiological reviews 78, 189-225, doi:10.1152/physrev.1998.78.1.189 (1998).
3. Hall, A., Provins, L. & Valade, A. Novel Strategies To Activate the Dopamine D1 Receptor: Recent Advances in Orthosteric Agonism and Positive Allosteric Modulation. J Med Chem 62, 128-140, doi:10.1021/acs.jmedchem.8b01767 (2019).
4. Kang, Y. et al. Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553-558, doi:10.1038/s41586-018-0215-y (2018).
